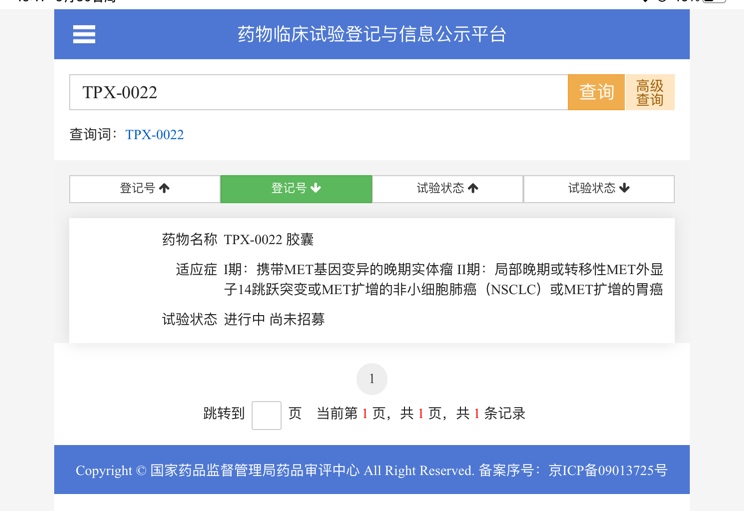
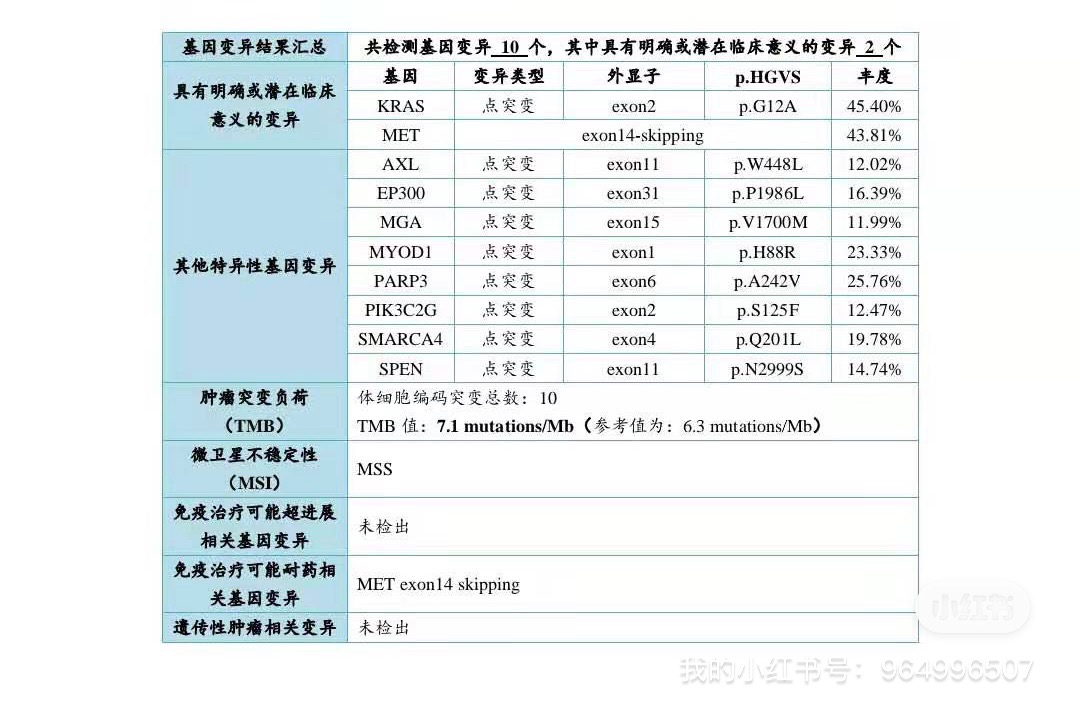
克唑替尼耐药不仅仅有1228 或1230, 而且可能混杂1200 1195等位置突变,这些突变对二型METTKI是不利的,因此上280克服这些1200 1195 突变后,再上二型会更好。
不过1228N对二型敏感度还是不行。
A unique finding for MET mutation-mediated resistance is that the D1228 and Y1230 resistance mutations are located in the A-loop in contrast to the gatekeeper, solvent front, and binding site mutations implicated in resistance to other tyrosine kinase inhibitors (38). The molecular basis of resistance of these unique A-loop mutations is evident from crystal structures of type I inhibitors such as crizotinib and capmatinib but not type II inhibitors including glesatinib, which bind independently of A-loop interactions. Moreover, molecular modeling of glesatinib bound to MET and the ability to adopt two distinct binding modes provides a molecular explanation as to why glesatinib retains activity against mutations within the A-loop and why the onset of resistance to glesatinib was delayed compared with type I MET inhibitors. Interestingly, the MET L1195V and F1200L/I mutations that were associated with resistance to both inhibitor classes in MET-mutant variant enzyme screens and/or resistance studies occur distal from the ligand binding site of type I inhibitors and may result in enhanced substrate affinity or destabilization of the autoinhibited A-loop conformation. Consistent with the observed resistance, biochemical screens in the current studies indicated a 10-fold loss of activity for crizotinib for MET F1200I compared with WT and this was the same amino acid that was found mutated in a crizotinib drug resistance screen (39). Although the MET F1200I mutation did not emerge as an acquired resistance mechanism during glesatinib drug resistance screens, there is direct involvement of this residue in the type II DFG-out binding mode, which would provide a clear molecular basis for attenuated activity for glesatinib and other type II inhibitors and is consistent with the 4-fold decrease in potency in crizotinib-resistant cells that harbored the Y1230H/F1200L double mutation. Interestingly, the type Ib inhibitor, savolitinib, demonstrated potent inhibitory activity against the F1200I variant suggesting it has a binding mode distinct from other type Ib MET inhibitors and supports the utility of this agent as another potential option to combat mutation mediated resistance via sequential treatment strategies. The relatively modest loss of glesatinib activity against the L1195V and F1200L mutations relative to most other type I inhibitors suggests that glesatinib may not rely solely on a single binding mode and may be able to adopt a conformation that attenuates loss of activity. Glesatinib was also evaluated against a panel of twelve other known MET mutations and activity was comparable with WT MET; thus, no additional anticipated glesatinib resistance mutations have been identified (partial data summary in Supplementary Table S2). |
 希望给没有阅读过的人一些体会(8)
本文来自于此书,只是做一些摘抄,分享给深陷迷茫的人。文中观点真实与否,效果怎
希望给没有阅读过的人一些体会(8)
本文来自于此书,只是做一些摘抄,分享给深陷迷茫的人。文中观点真实与否,效果怎
 盲试靶向药,第36个月,这一次无效
过完年以后因为工作调用原因,一只没有更新帖子,很多关心我的病友也都发信息问我们最
盲试靶向药,第36个月,这一次无效
过完年以后因为工作调用原因,一只没有更新帖子,很多关心我的病友也都发信息问我们最
 公公婆婆接连确诊肺癌,我们很后悔没
讲述者:小小整理者:pear
清晨的阳光,温柔地抚上窗棂。闹钟轻响,新的一天在熟悉的
公公婆婆接连确诊肺癌,我们很后悔没
讲述者:小小整理者:pear
清晨的阳光,温柔地抚上窗棂。闹钟轻响,新的一天在熟悉的
 老公刚刚54岁,确诊肺癌,手术中发现
各位朋友好。
度过了慌乱的一个月,有机会发现了这个论坛,如获至宝,说说我家
老公刚刚54岁,确诊肺癌,手术中发现
各位朋友好。
度过了慌乱的一个月,有机会发现了这个论坛,如获至宝,说说我家
 历经3次靶向治疗耐药,如今妈妈也成
作者:浅斟低唱
妈妈确诊肺癌,医生为我们指明治疗方向第一阶段:(2017.4-2019.5,24
历经3次靶向治疗耐药,如今妈妈也成
作者:浅斟低唱
妈妈确诊肺癌,医生为我们指明治疗方向第一阶段:(2017.4-2019.5,24





 显身卡
显身卡